









Stability Studies: Pharmaceutical Drug Products: As per FDA and ICH
- Moral Randeria

- Aug 1, 2025
- 8 min read
Updated: Sep 16, 2025
Introduction
Stability studies are important for verifying the quality, safety, and effectiveness of drug products during their shelf life. Stability studies reveal the physical, chemical, biological, and microbiological changes that may happen in a drug product under different environmental conditions, such as temperature, humidity, light, and oxygen. Stability studies also help to determine the storage conditions and expiry dates of drug products.
The FDA and the ICH have given guidance on how to plan, execute, and report stability studies for drug products. The purpose of these guidelines is to align the regulatory standards and requirements for stability studies in different regions and countries, and to support the global development and registration of drug products. The FDA and ICH guidelines address various topics of stability studies, such as the kinds of studies, the test methods, the parameters that indicate stability, the acceptance criteria, the analysis of stability data, and the presentation of stability data.
Types of Stability Studies
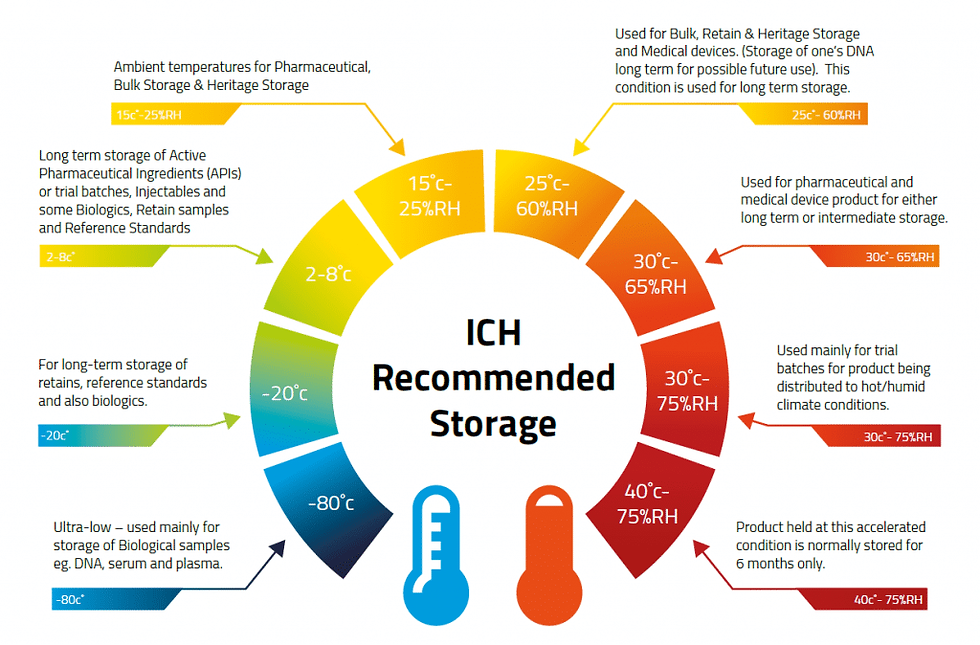
There are four main types of stability studies that the FDA and ICH guidelines recognize: long-term, accelerated, intermediate, and stress studies. The table below shows the duration and objective of each type of study.
· Long-term studies: These show the main stability of a drug product under the suggested storage conditions. Long-term studies use the expected or planned storage temperature and humidity for the whole shelf life of the drug product. For example, for a drug product with a 24-month shelf life, long-term studies last for 24 months or more.
· Speeded up studies: These are the extra stability studies that give more information on how stable a drug product is under harsher conditions than the advised storage conditions. Speeded up studies are done at higher temperatures and/or higher relative humidities than the long-term studies, and for a shorter time. The aim of speeded up studies is to see how temperature and humidity affect the stability of a drug product, and to estimate the shelf life of the drug product based on the speeded-up data. For example, for a drug product with a suggested shelf life of 24 months, speeded up studies are done for 6 months at 40°C and 75% relative humidity.
· Intermediate studies: Intermediate studies are optional stability studies that supplement the information on how stable a drug product is under conditions that fall between the long-term and accelerated conditions. Intermediate studies are done at intermediate temperatures and/or relative humidities, and for a time period that is between the long-term and accelerated studies. The aim of intermediate studies is to verify the stability of a drug product under the suggested storage conditions, and to support the extension of the shelf life beyond the long-term data. For instance, for a drug product with a proposed shelf life of 24 months, intermediate studies are carried out for 12 months at 30°C and 65% relative humidity.
· Stress studies: These are the experiments that test the stability of a drug product under harsh conditions of temperature, humidity, light, oxidation, pH, and mechanical stress. Stress studies are done at higher or lower temperatures and/or relative humidities than the accelerated studies, and for a shorter time. The aim of stress studies is to find out the possible degradation products and ways of a drug product, and to set up the parameters and methods that indicate the stability of the drug product. For example, for a drug product, stress studies are performed for a few days or weeks at 50°C and 80% relative humidity, or under exposure to light, oxygen, acid, or base.
Test Methods and Parameters
The FDA and ICH guidelines state that the test methods and parameters for stability studies must be stability-indicating, validated, and specific for the drug product. Stability-indicating methods and parameters can identify any changes in the drug product's identity, strength, quality, purity, and potency caused by degradation or interaction with the container-closure system.
Validated methods and parameters have been confirmed to be accurate, precise, sensitive, specific, robust, and reproducible for the drug product. Specific methods and parameters can assess the drug product without being affected by the excipients, impurities, or degradation products. The methods and factors for testing stability may differ based on the kind, form, and ingredients of the drug product.
However, some of the typical methods and factors for testing stability are:
· Appearance: This is how the drug product looks, such as its color, clarity, shape, size, and consistency. Appearance is checked by using a light and a magnifying glass or a microscope.
· Assay: This is the numerical measure of how much or how concentrated the active ingredient or the drug substance is in the drug product. Assay usually uses a chromatographic, spectroscopic, or titrimetric method.
· Content uniformity: This is the measurement of the uniform distribution of the active ingredient or the drug substance in the drug product. Content uniformity is usually assessed by using a statistical method, such as the acceptance value (AV) or the coefficient of variation (CV).
· Dissolution: This measures how fast and how much of the drug substance or active ingredient comes out of the drug product into a liquid. Dissolution usually uses a standard device, like a basket, a paddle, or a flow-through cell, and a proper liquid, like water, buffer, or fake stomach or intestine fluid.
· Impurities: These are the substances that may be found in the drug product that are not desired or intended. They may come from the synthesis, manufacture, storage, or degradation of the drug product. Contaminants may consist of organic contaminants, such as related compounds, breakdown products, or residuals, or inorganic contaminants, such as heavy metals, or elemental contaminants. Contaminants are typically quantified by using a method based on chromatography, spectroscopy, or wet chemistry.
· Moisture content: This is the measurement of the amount of water or moisture that may be present in the drug product due to the hygroscopicity of the drug substance or the excipients, or due to the exposure to humidity during storage. Moisture content is usually measured by using a gravimetric, titrimetric, or spectroscopic method.
· pH: This is the indicator of how acidic or basic the drug product is, especially for dosage forms that are liquid or semi-solid. pH is usually checked by using a normal pH meter or a pH marker.
· Microbial limits: These are the values of the number and kind of microorganisms that can be found in the drug product because of the contamination during the production, packing, or storage of the drug product.
Microbial limits consist of the total aerobic microbial count (TAMC), the total combined yeast and mold count (TYMC), and the lack of specific pathogens, such as Escherichia coli, Salmonella, Staphylococcus aureus, and Pseudomonas aeruginosa. Microbial limits are usually determined by using a standard plate count, a membrane filtration, or a rapid microbiological method.
Acceptance Criteria
According to the FDA and ICH guidelines, the specifications, the label claims, and the clinical relevance of the drug product should determine the acceptance criteria for stability studies.
Specifications are the numerical limits, ranges, or other criteria that set the acceptable quality of the drug product. Label claims are the statements or information that indicate the identity, strength, quality, purity, and potency of the drug product.
Clinical relevance is the effect or importance of the stability data on the safety and efficacy of the drug product. The acceptance criteria for stability studies may differ depending on the kind, dosage form, and composition of the drug product.
Some of the general guidelines for establishing the acceptance criteria for stability studies are:
· The assay should meet the acceptance criteria of being within ±5% of the label claim, unless there is a valid reason otherwise.
· The content uniformity should meet the acceptance criteria of being within the range of 85% to 115% of the label claim, unless there is a valid reason otherwise.
· The dissolution should meet the acceptance criteria of being within ±10% of the label claim, unless there is a valid reason otherwise.
· The impurity limits should follow the relevant pharmacopoeia (e.g., USP, EP, or JP) or ICH guidelines (e.g., Q3A, Q3B, or Q3D), unless there is a good reason not to.
· The moisture content limits should follow the relevant pharmacopoeia or depend on the stability data and the clinical importance of the drug product.
· The acceptance criteria for pH should be within the range specified in the relevant pharmacopoeia or based on the stability data and the clinical relevance of the drug product.
· The microbial limits should meet the criteria specified in the relevant pharmacopoeia or be based on the stability data and the clinical significance of the drug product.
Stability Data Analysis
The FDA and ICH guidelines suggest that the stability data analysis should be done by using suitable statistical methods, such as regression analysis, analysis of variance (ANOVA), or shelf life estimation. The stability data analysis aims to assess the patterns, variation, and importance of the stability data, and to calculate the shelf life of the drug product based on the stability data.
How to analyze the stability data may differ based on the drug product's kind, form, and ingredients.
However, some of the common methods for doing the stability data analysis are:
· The stability data analysis should be done on each batch of the drug product, and not on the combined or mean data, unless there is a good reason.
· The stability data analysis should be done on the original units of the stability data, and not on the changed or adjusted data, unless there is a good reason.
· The stability data analysis should be done on the most extreme case of the stability data, such as the lowest assay, the highest impurity, or the lowest dissolution, unless there is a good reason.
· Use a linear or nonlinear regression model to analyze the stability data, based on the data type and behavior, and validate the model with suitable criteria, such as R2, RSS, or AIC.
· Use α = 0.05 and (1-α) = 0.95 to analyze the stability data, unless you have a good reason not to.
· Use a shelf life estimation method, such as the intercept method, the slope method, or the Q1 method, to analyze the stability data, based on the data type and behavior, and justify the method with sound logic.
Stability Data Presentation
The FDA and ICH guidelines advise that the stability data presentation should be transparent, brief, and coherent, and should contain all the pertinent information and documentation of the stability studies. The aim of the stability data presentation is to convey the findings, implications, and suggestions of the stability studies to the regulatory authorities and the stakeholders.
The drug product's type, dosage form, and composition may affect how the stability data is presented.
However, some of the usual parts of the stability data presentation are:
· The stability studies' summary, including the purpose, design, methods, parameters, criteria, analysis, and results.
· The stability data's table, including the batch number, size, date of manufacture and expiry, storage condition, test interval, method, parameter, and result for each batch of the drug product.
· The stability data's graph, including the x-axis, y-axis, legend, title, and label for each graph of the stability data.
· A summary of the stability data, including how they were interpreted, evaluated, and compared, and how any unusual or inconsistent results were explained.
· A decision on the stability data, including how the shelf life of the drug product was justified, validated, and extrapolated based on the data.
· A suggestion of the stability data, including the storage conditions, expiration dates, and labels of the drug product based on the data.
· A source of the stability data, including the mention and recognition of any materials, guidelines, or standards that were applied or followed in the stability studies.
References
· FDA. (2018). Guidance for Industry: Q1A(R2) Stability Testing of New Drug Substances and Products.
· FDA. (2014). Guidance for Industry: Q1E Evaluation of Stability Data.
· ICH. (2003). ICH Harmonised Tripartite Guideline: Q1A(R2) Stability Testing of New Drug Substances and Products.
· ICH. (2003). ICH Harmonised Tripartite Guideline: Q1E Evaluation of Stability Data.
· ICH. (2006). ICH Harmonised Tripartite Guideline: Q1F Stability Data Package for Registration Applications in Climatic Zones III and IV.
· ICH. (1996). ICH Harmonised Tripartite Guideline: Q3A(R2) Impurities in New Drug Substances.
· ICH. (2006). ICH Harmonised Tripartite Guideline: Q3B(R2) Impurities in New Drug Products.
· ICH. (2014). ICH Harmonised Guideline: Q3D Elemental Impurities.
· USP. (2019). General Chapter Validation of Compendial Procedures.
· USP. (2019). General Chapter Uniformity of Dosage Units.
· USP. (2019). General Chapter Dissolution.
· USP. (2019). General Chapter Water Determination.
· USP. (2019). General Chapter Microbiological Examination of Nonsterile Products: Acceptance Criteria for Pharmaceutical Preparations and Substances for Pharmaceutical Use.


Comments